4 归一化
本文主要解释了为什么要做归一化,各种归一化的方法解释及R语言怎么做,并附上了一个在线做归一化的网址(http://bioladder.cn)。
4.1 为什么要做归一化?
在生物学实验中,往往会因为上样量等差别,导致本来实际样本中相似的定量值的结果因为实验原因导致有整个样本的整体偏差。因此,归一化的目的是把实验导致的整体偏差校正回来。
4.2 各种归一化的方法解释及R语言怎么做
归一化方法有很多,主要包括:中值归一化、共有蛋白中值归一化、线性函数归一化、Z Score 归一化等。选择哪种归一化方法需要根据具体数据的情况,结合归一化方法的原理判断。
4.2.1 中值归一化
即每列数据除以该列数据的中位数。采用中值归一化的前提假设是:需要比对的多个样本中,大部分蛋白的表达量是相似的。而中位数是大部分蛋白所集中的点,受该样本的极大值或极小值定量影响很小,把每个样本的定量中值归一化成一个相等的值,可以有效地保证归一化后,大部分蛋白的表达量区间相似。
# 读取原始数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/normalize/demo.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
row.names = 1 # 指定第一列为行名,根据实际数据修改
)
# 中位数归一化
nor_median=function(x){
y=na.omit(x)
return(x/median(y))
}
dfmedian = apply(df, 2,nor_median) # 2是对列进行计算;1是对行进行计算4.2.2 共有蛋白中值归一化
即每列数据除以共有蛋白的中位数。这种方法依然是假设大部分蛋白表达量相似,但碰到部分特殊结果,也即部分样本鉴定数量少地多(比如只有其它样本的1/2甚至更少),直接用中值归一化会导致整体的偏移,因此只用共有蛋白做归一化。
# 共有蛋白中值归一化
library(tidyverse)
rmnaMedianNormalize = function(x,df){
median = median(x[complete.cases(df)])
x/median
}
dfrmnaMedian = df %>%
mutate(across(
where(is.numeric),
~rmnaMedianNormalize(.x,df)
))4.2.3 线性函数归一化
是对原始数据的线性变换,使结果值映射到[0-1]之间,保留源数据存在的关系,消除取值范围最简单的方法,不过,极易受个别离群值影响,如果数据集中某个数值很大,其他各值归一化后会接近0。这种方法的假设是,各个样本中,最大值和最小值都是相似的
# 线性函数归一化
nor_min_max=function(x){
y=na.omit(x)
return((x - min(y))/(max(y) - min(y)))
}
dfmin_max = apply(df, 2,nor_min_max) # 2是对列进行计算;1是对行进行计算4.2.4 Z Score 归一化
经过处理的数据符合标准正态分布,即均值为0,标准差为1。这种方法的假设是,每个样本的定量值服从正态分布,大部分蛋白表达量相似,且方差也相似
dfZScore = scale(df)4.2.5 最大值归一化
前提假设是,每个样本的最大值蛋白相似,且定量较稳定。用得不多。单靠一个最大值,变异度较大。
nor_max=function(x){
y=na.omit(x) # 去除空值
return(x/max(y))
}
dfmax = apply(df, 2,nor_max) # 2是对列进行计算;1是对行进行计算4.2.6 总量归一化
前提假设是,每个样本蛋白总量相似,因此归一化成总量一致。这种方法用得比较多,包括RNA-seq中常用的RPKM等。
# 总和归一化
nor_sum=function(x){
y=na.omit(x)
return(x/sum(y))
}
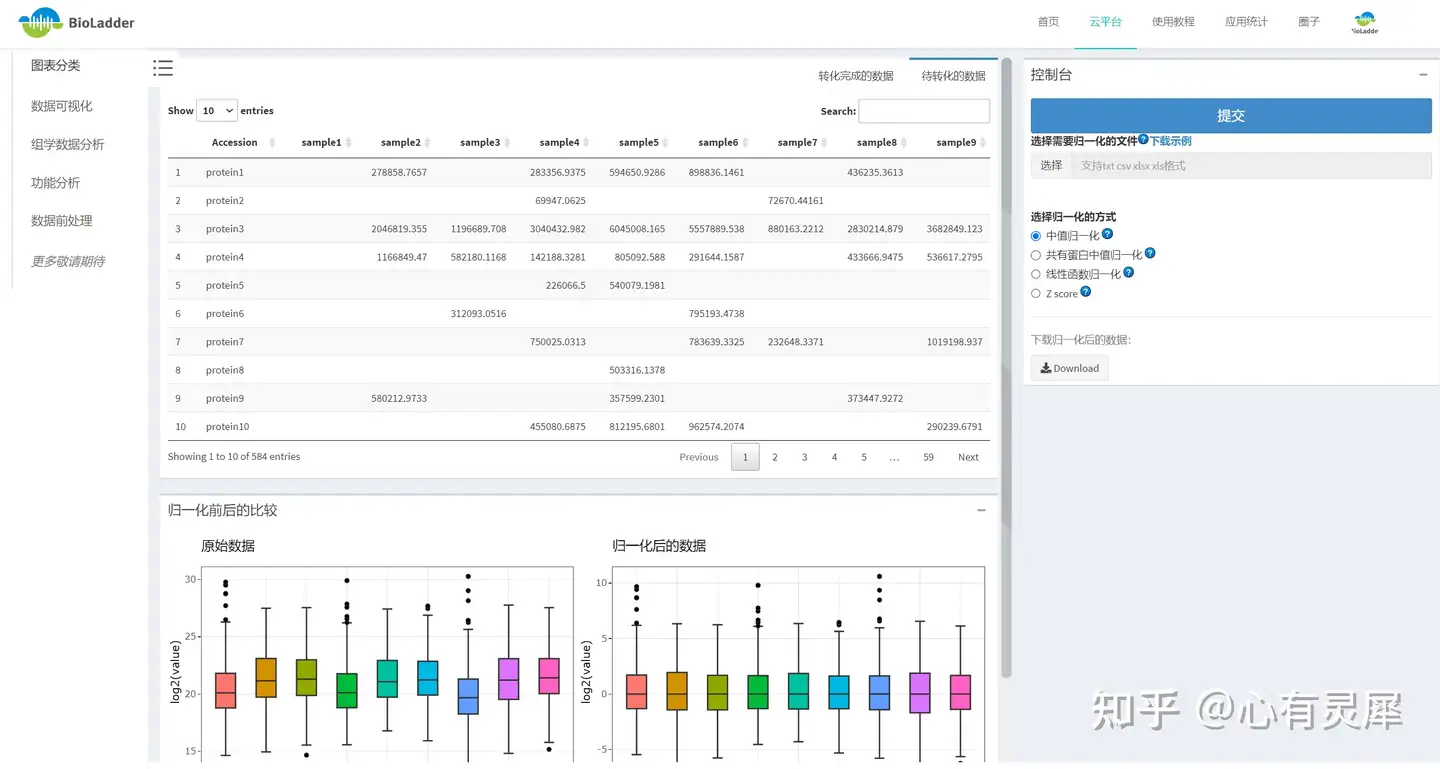
dfsum = apply(df, 2,nor_sum) # 2是对列进行计算;1是对行进行计算4.2.7 BioLadder云平台免费在线做归一化
不想写代码?可以用BioLadder生信云平台在线做归一化。
网址:
归一化-BioLadder生物信息在线分析可视化云平台www.bioladder.cn/web/#/chart/48