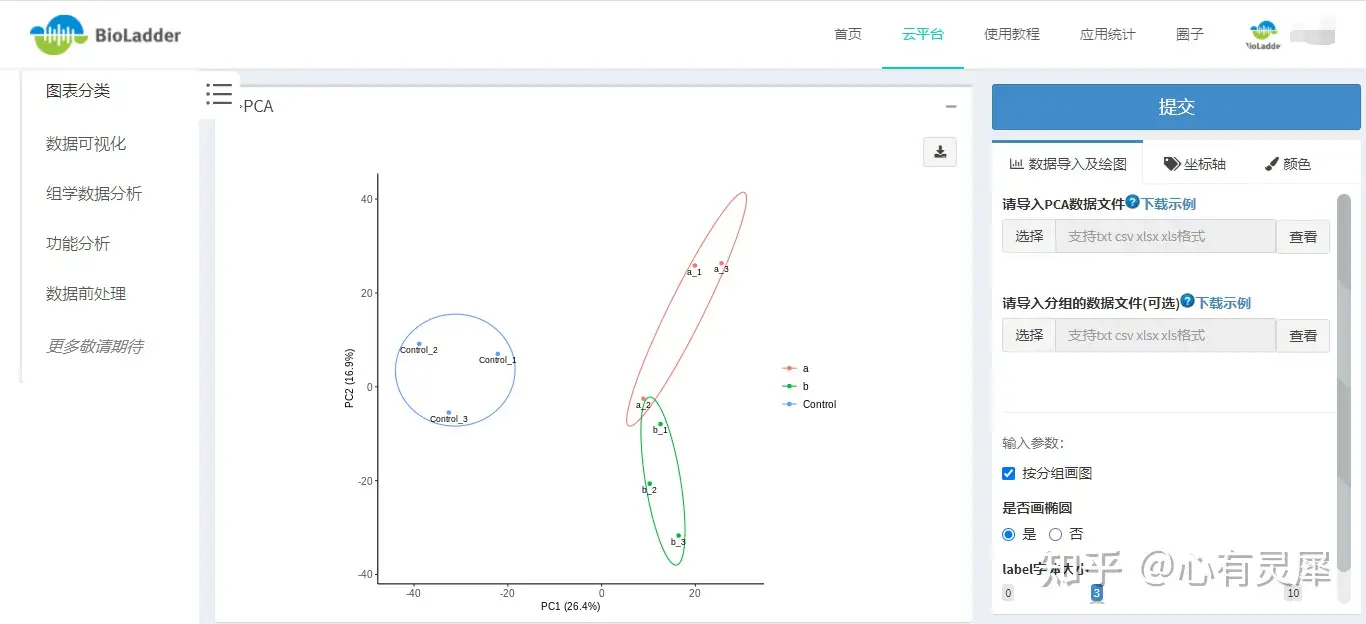
12 PCA
12.1 什么是PCA?
人眼一般能感知的空间为二维和三维。高维数据可视化的重要目标就是将高维数据呈现于二维或三维空间中。高维数据变换就是使用降维度的方法,使用线性或非线性变换把高维数据投影到低维空间,去掉冗余属性,但同时尽可能地保留高维空间的重要信息和特征。
主成分分析法,也被称为主分量分析法,是很常用的一种数据降维方法。主成分分析法采用一个线性变换将数据变换到一个新的坐标系统,使得任何数据点投影到第一个坐标(第一主成分)的方差最大,在第二个坐标(第二主成分)的方差为第二大,以此类推。因此,主成分分析可以减少数据的维数,并保留对方差贡献最大的特征。
本文我们就来讨论一下PCA图是如何绘制的以及如何对其进行解读。

12.2 绘图前的数据准备
demo数据可以在https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/PCA.rar下载。
12.2.1 PCA数据
数据来源一般是搜库结果定量表。包含2个维度的数据,一般情况下,每一行是一个基因,每一列是一个样本。
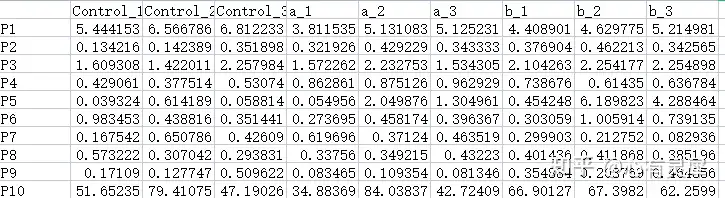
12.2.2 分组数据(可选)
行名的名称和个数要和之前的PCA数据保持一致,列名为分组名称,可以包含不止一个分组。

12.3 R语言怎么画PCA
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(ggbiplot)
# ggbiplot包需要从github上下载
# install.packages("devtools")
# library(devtools)
# install_github("vqv/ggbiplot")
# 读取PCA数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/data.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
)
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/PCA/sample.class.txt",
header = T,
row.names = 1
)
# PCA计算
pca_result <- prcomp(df,
scale=T # 一个逻辑值,指示在进行分析之前是否应该将变量缩放到具有单位方差
)
# 绘图
ggbiplot(pca_result,
var.axes=F, # 是否为变量画箭头
obs.scale = 1, # 横纵比例
groups = dfGroup[,1], # 添加分组信息,为分组文件的第一列
ellipse = T, # 是否围绕分组画椭圆
circle = F)+
geom_text( # geom_text一个在图中添加标注的函数
aes(label=rownames(df)), # 指定标注的内容为数据框df的行名
vjust=1.5, # 指定标记的位置,vjust=1.5 垂直向下1.5个距离。 负数为位置向上标记,正数为位置向下标记
size=2 # 标记大小
)
# 更多请 ?ggbiplot 获取帮助12.4 BioLadder生信云平台在线绘制PCA图
不想写代码?可以用BioLadder生信云平台在线绘制热图。
免费使用,注册登录后畅享40+模块。
网址:https://www.bioladder.cn/web/#/chart/13