16 NMDS分析
16.1 什么是NMDS分析?
人眼一般能感知的空间为二维和三维。高维数据可视化的重要目标就是将高维数据呈现于二维或三维空间中。高维数据变换就是使用降维度的方法,使用线性或非线性变换把高维数据投影到低维空间,去掉冗余属性,但同时尽可能地保留高维空间的重要信息和特征。
非度量多维标度(NMDS)分析,是PCoA的非度量替代方法。NMDS是一种将多维空间的研究对象(样本或变量)简化到低维空间进行定位、分析和归类,同时又保留对象间原始关系的数据分析方法。NMDS需要在一开始就要指定维度(轴)的数量,如预设2-3个排序轴,计算过程中将根据已定义好的轴数分配对象坐标。而PCoA则基于特征向量提取,维度(轴)的数量由数据集的固有属性决定(对象数-1),获得样方排序后再根据特征值等信息自定义确定选择的轴数。
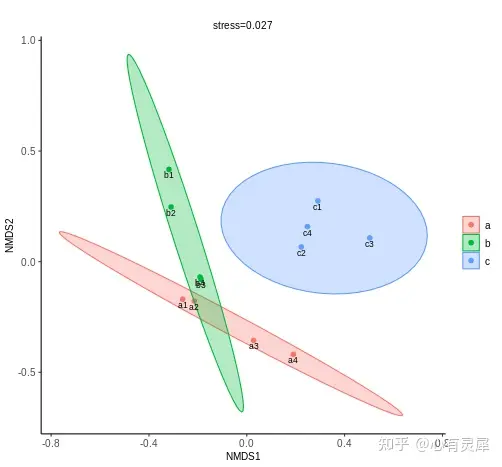
本文我们就来讨论一下如何做NMDS分析以及如何对其进行解读。
16.2 分析前的数据准备
需要2个文件,demo数据可以在https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/NMDS/NMDS.zip下载。
16.2.1 NMDS数据
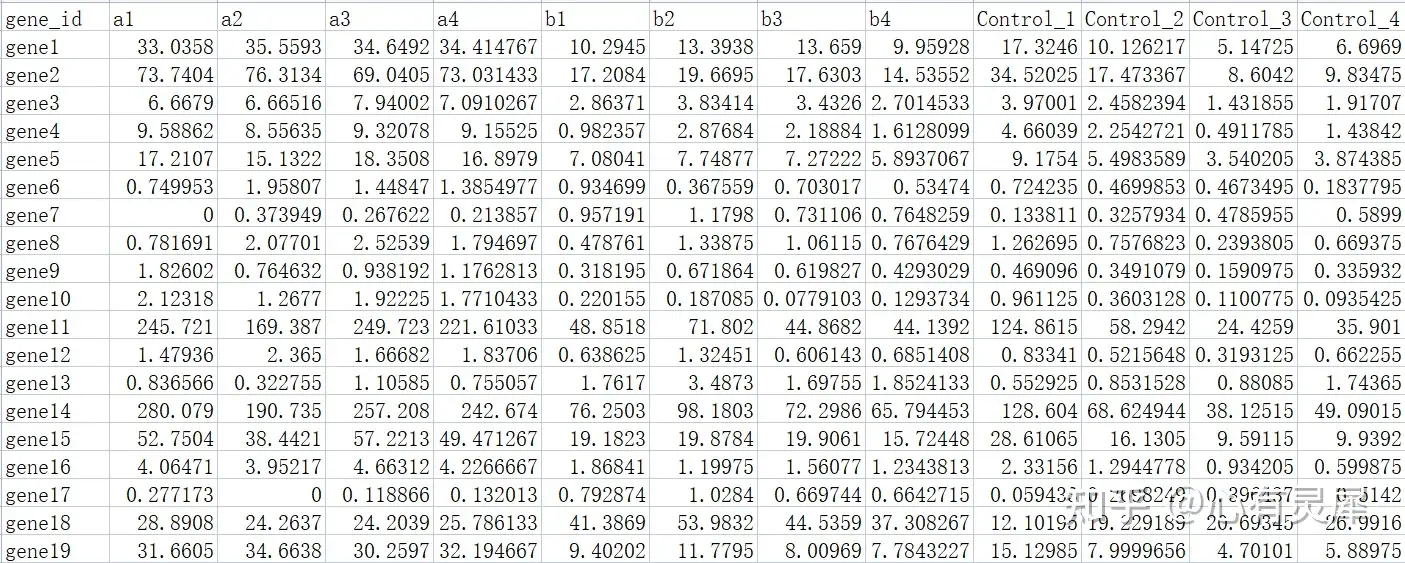
数据来源一般是搜库结果定量表。包含2个维度的数据,一般情况下,每一行是一个基因,每一列是一个样本。
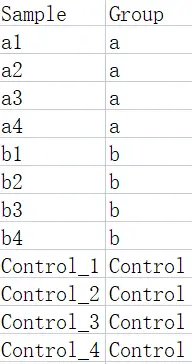
16.2.2 分组数据(可选)
行名的名称和个数要和之前的NMDS数据保持一致,列名为分组名称,可以包含不止一个分组。
16.3 R语言怎么画NMDS
library(ggplot2)
library(vegan)
# 读取NMDS数据文件
df = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/NMDS/1.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
)
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.bioladder.cn/shiny/zyp/bioladder2/demoData/NMDS/2.txt",row.names = 1)
# NMDS计算
dfNmds<-metaMDS(df,distance="bray",k = 2)
# 绘图前的数据整理
data = data.frame(dfNmds$points)
data$group = dfGroup$Group
# 绘图
ggplot(data,aes(x = MDS1,
y = MDS2,
color = group,
group = group,
fill = group)
)+
geom_point(size=2)+
theme_classic()+
stat_ellipse( # 添加置信区间
geom = "polygon",
level = 0.95,
alpha=0.3)+
geom_text( # 添加文本标签
aes(label=rownames(data)),
vjust=1.5,
size=2,
color = "black"
)+
labs( # 在副标题处添加stress
subtitle = paste("stress=",round(dfNmds$stress,3),sep="")
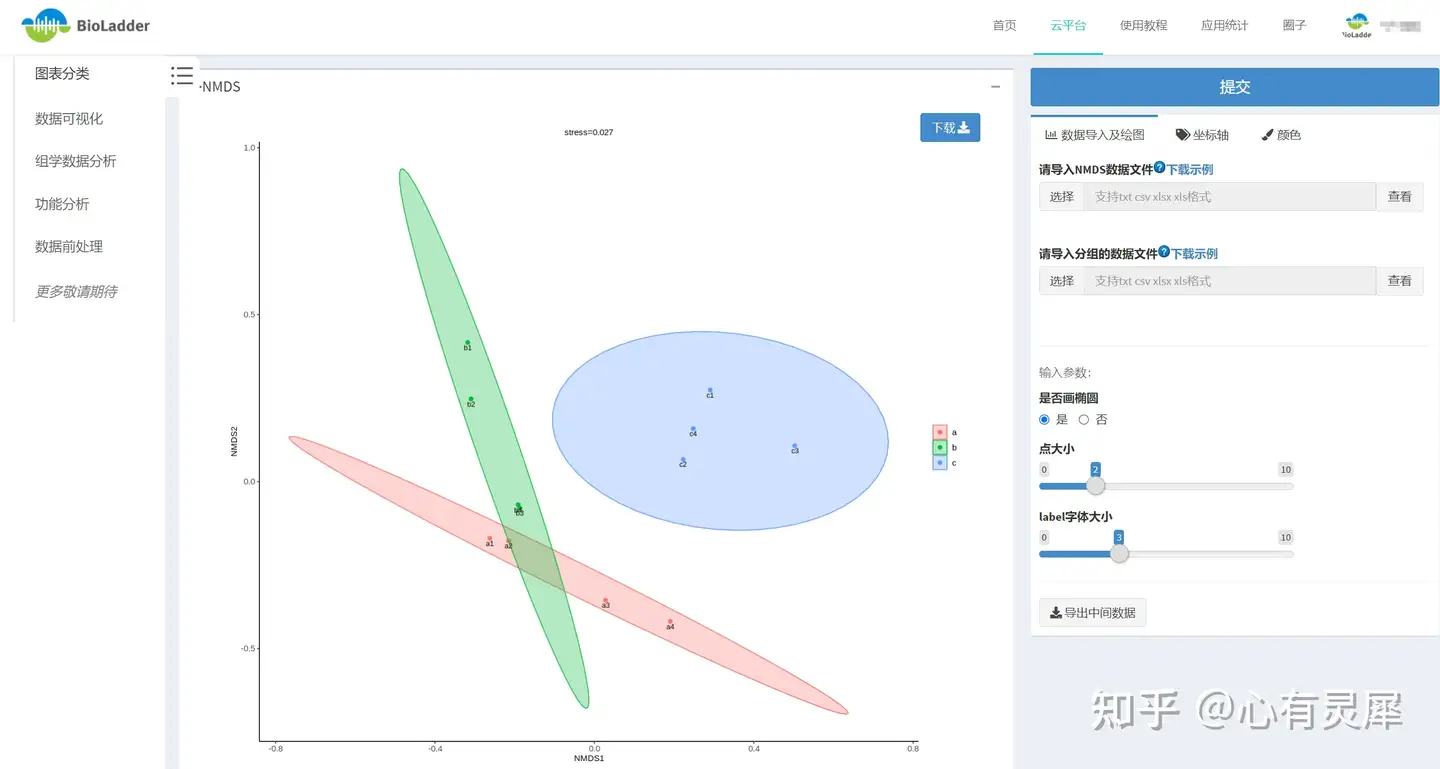
)16.4 BioLadder生信云平台在线做NMDS分析
不想写代码?可以用BioLadder生信云平台在线绘制NMDS。
网址:https://www.bioladder.cn/web/#/chart/71
16.5 NMDS结果解读
对不同样本间的差异程度,通过点与点间的距离体现的,最终获得样本的空间定位点图。