# A tibble: 5,810 × 5
X Group1 Group2 Group3 Group4
<chr> <dbl> <dbl> <dbl> <dbl>
1 Gene1 0.0396 0.0185 NA 0.00256
2 Gene2 0.0462 0.0326 0.0473 0.0200
3 Gene3 NA 0.0398 0.0234 NA
4 Gene4 0.125 0.556 0.284 0.253
5 Gene5 1.05 2.95 1.55 2.27
6 Gene6 0.0174 0.0101 NA NA
7 Gene7 0.00677 NA 0.00607 NA
8 Gene8 0.545 0.796 0.394 0.529
9 Gene9 0.544 1.89 1.03 1.20
10 Gene10 0.0140 0.00347 0.00385 0.0102
# ℹ 5,800 more rowsR语言如何做趋势分析
前言
本篇是R语言Mfuzz包做趋势分析的教程。
什么是趋势分析?
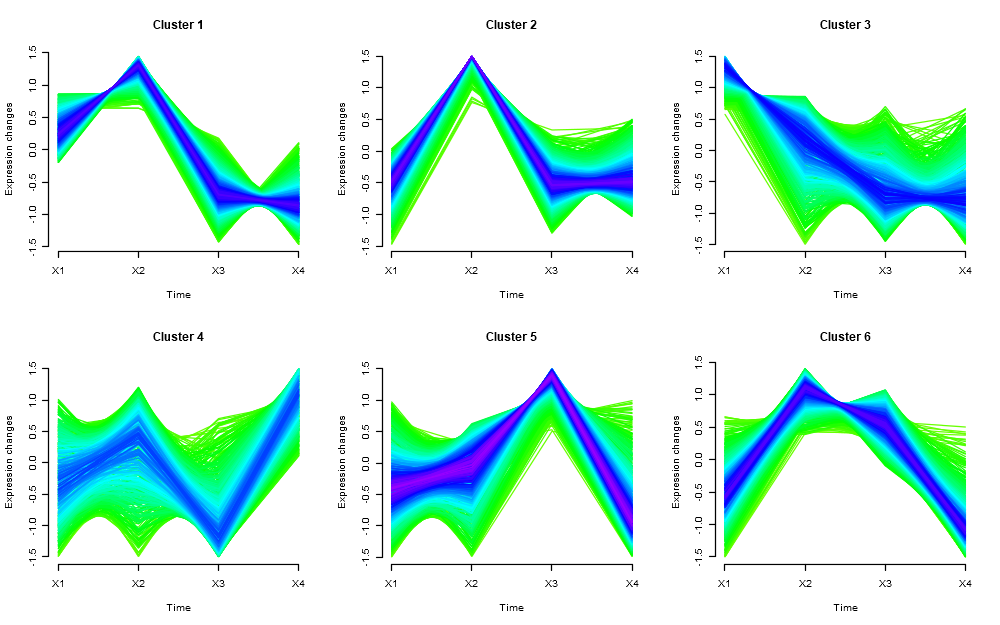
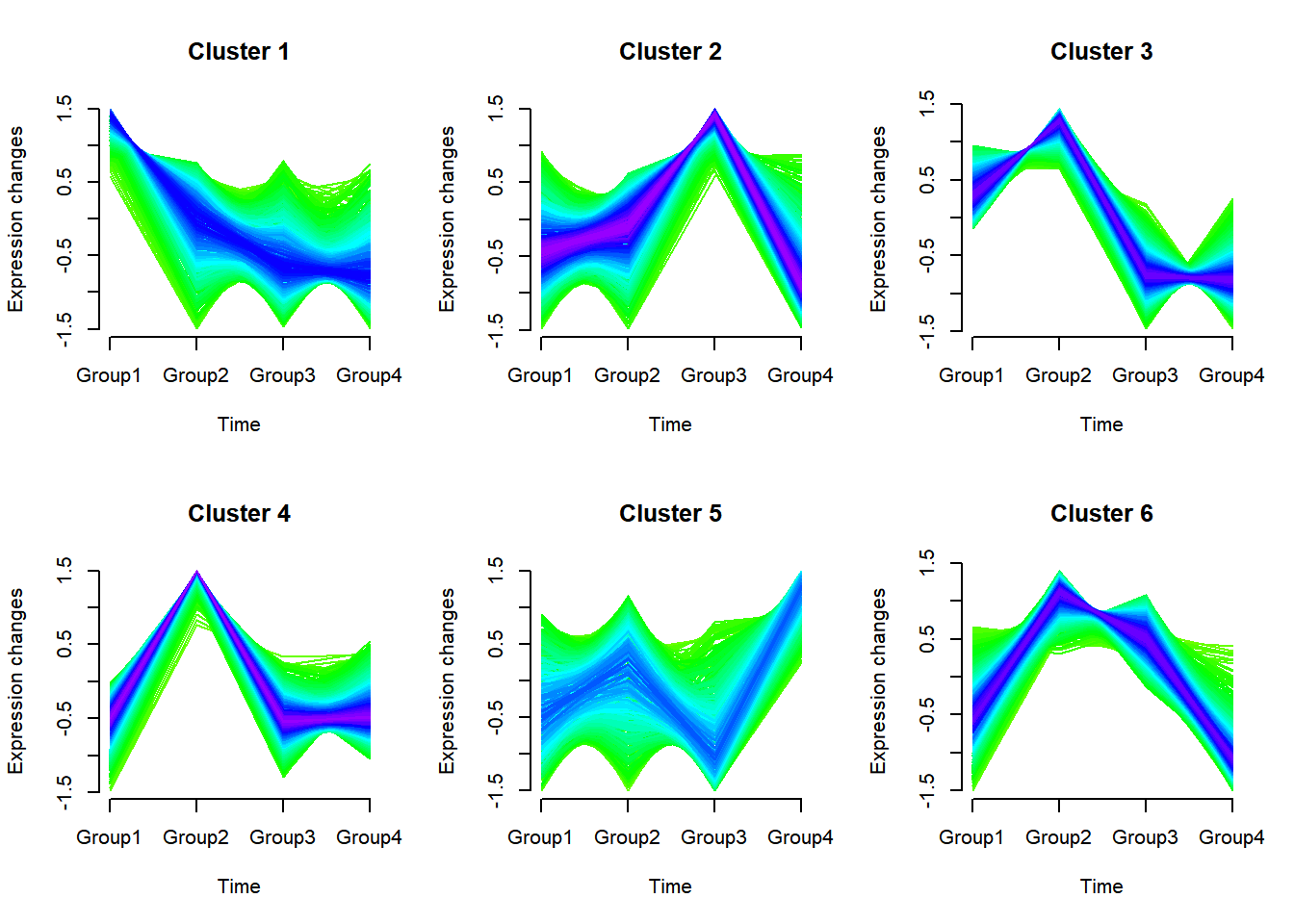
基于基因表达值进行了聚类,对于每个簇中的基因,具有相似的时间表达特征。随后,即可从图中识别一些重要的聚类簇,比方说簇中基因随时间表达趋势增加或减少,以及在特定时间出现了更高或更低的表达等,以建立和观察的表型的联系。
绘图前的数据准备
demo数据可以在https://www.r2omics.cn/res/demodata/mfuzz.txt下载。
数据包含2个维度,数据通常来源于搜库结果定量表,且需要有时序关系。一般都是用预处理后的数据,组内取平均值或中位数。
R语言如何绘制趋势分析聚类图
# 代码来源:https://www.r2omics.cn/
# Mfuzz包的安装方式为:BiocManager::install("Mfuzz")
library(Mfuzz)
# 读取时间序列分析数据文件
df = read.delim("https://www.r2omics.cn/res/demodata/mfuzz.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
row.names = 1
)
# 构建对象,填补缺失值,标准化等
dm <- data.matrix(df) # 数据框转换为矩阵
ESet <- new("ExpressionSet",exprs = dm) # 构建对象
ESet <- filter.NA(ESet, thres=0.25) # 过滤缺失值超过“25%”的基因1215 genes excluded.ESet <- fill.NA(ESet,mode="knn") # knn算法填补缺失值
ESet <- filter.std(ESet,min.std=0,visu=F) # 根据标准差去除样本间差异太小的基因0 genes excluded.gene.s <- standardise(ESet) # 标准化
# exprs(gene.s) # 查看处理后的数据
# 聚类
c <- 6 # 设置聚类个数
m <- mestimate(gene.s) # 评估出最佳的m值
set.seed(123) # 设置随机种子,防止每次聚类的结果都不一样,无法复现
cl <- mfuzz(gene.s, c = c, m = m)
# cl # 查看每个基因聚到哪个类当中
# cl$size # 查看每个cluster中的基因个数
# cl$membership # 查看基因和cluster之间的membership。基因在哪个cluster中的membership越高,基因就越会划分到这类。
# 绘图
mfuzz.plot(
gene.s,
cl,
time.labels = colnames(df), # 设置x轴显示的名称为原数据的列名
mfrow=c(2,3), # 图形排列方式,2行3列
new.window= FALSE) # 是否在新窗口打开