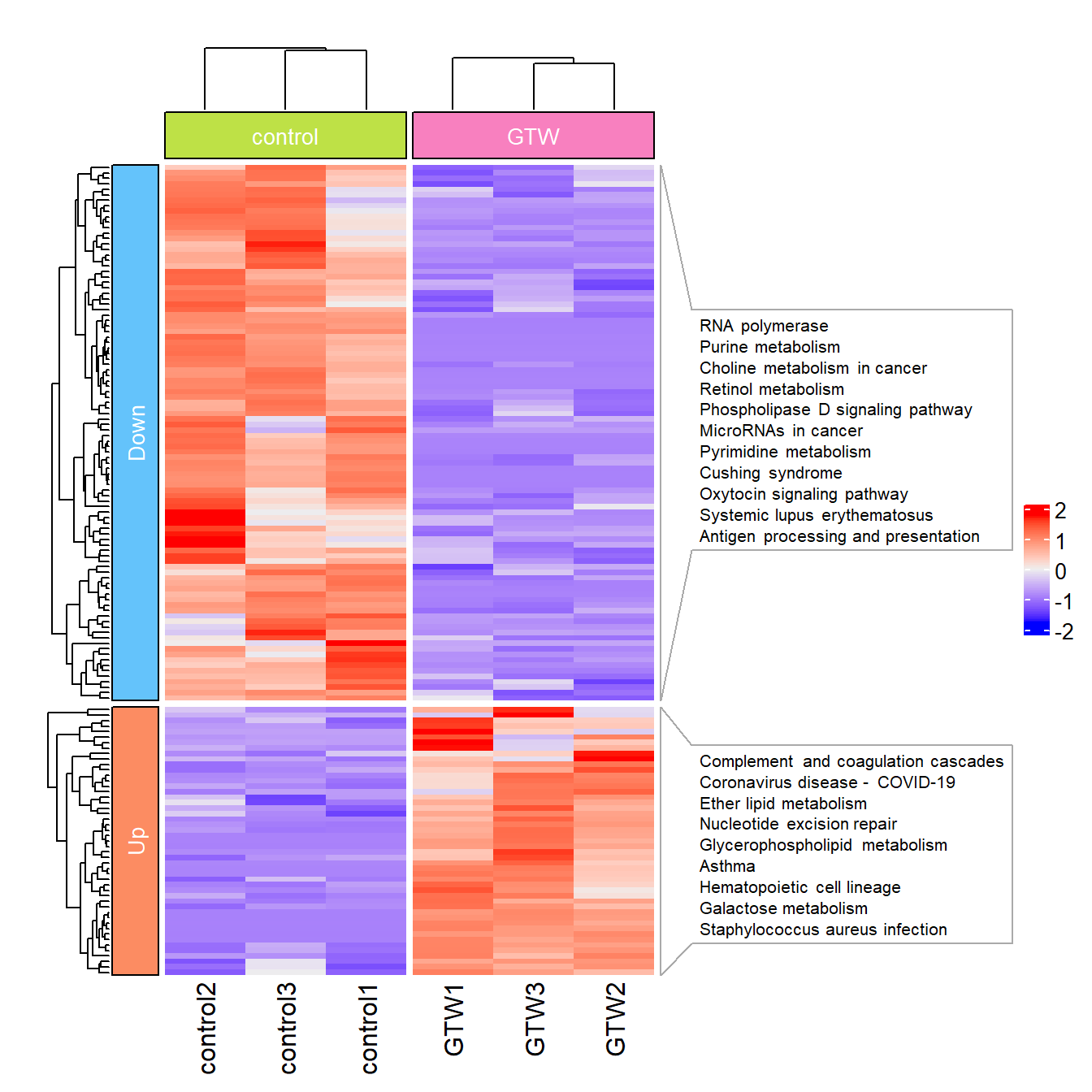
# 代码来源:https://www.r2omics.cn/
# 加载必要的R包
library(ComplexHeatmap)
library(dplyr)
# 读取热图数据文件(数据文件路径需要根据实际情况进行调整)
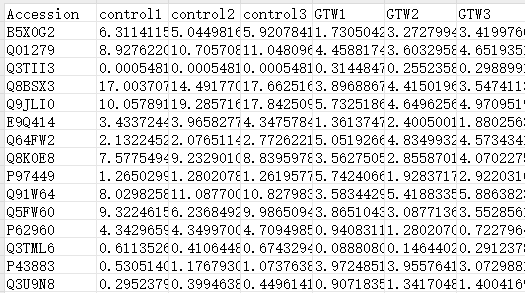
df = read.delim("https://www.r2omics.cn/res/demodata/heatmapAnno/data.txt", sep = "\t", row.names = 1)
# 读取分组富集数据文件
# 分组文件里面的顺序应该和数据文件保持一致
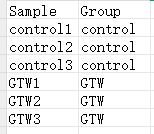
dfSample = read.delim("https://www.r2omics.cn/res/demodata/heatmapAnno/dfSample.txt", row.names = 1, sep = "\t") # 样本分组信息
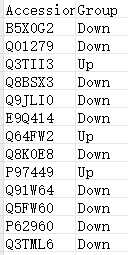
dfGene = read.delim("https://www.r2omics.cn/res/demodata/heatmapAnno/dfGene.txt", row.names = 1, sep = "\t") # 基因分组信息
dfEnrich = read.delim("https://www.r2omics.cn/res/demodata/heatmapAnno/dfEnrich.txt", sep = "\t") # 富集条目数据
# 把数据按行进行Z-score归一化
df.scale = df %>%
t() %>% # 转置矩阵,使得每一列为一个样本
scale() %>% # 对每一列进行Z-score标准化
t() %>% # 再次转置回原来的矩阵结构
data.frame(check.names = FALSE) # 转换为数据框
# 整理富集条目数据,根据分组生成每个组的相关富集条目
dfsentences = dfEnrich %>%
group_by(Group) %>% # 按照组别分组
summarise(Terms = list(Term)) %>% # 获取每个组的富集条目
pull(Terms, name = "Group") # 提取富集条目数据,按组名命名
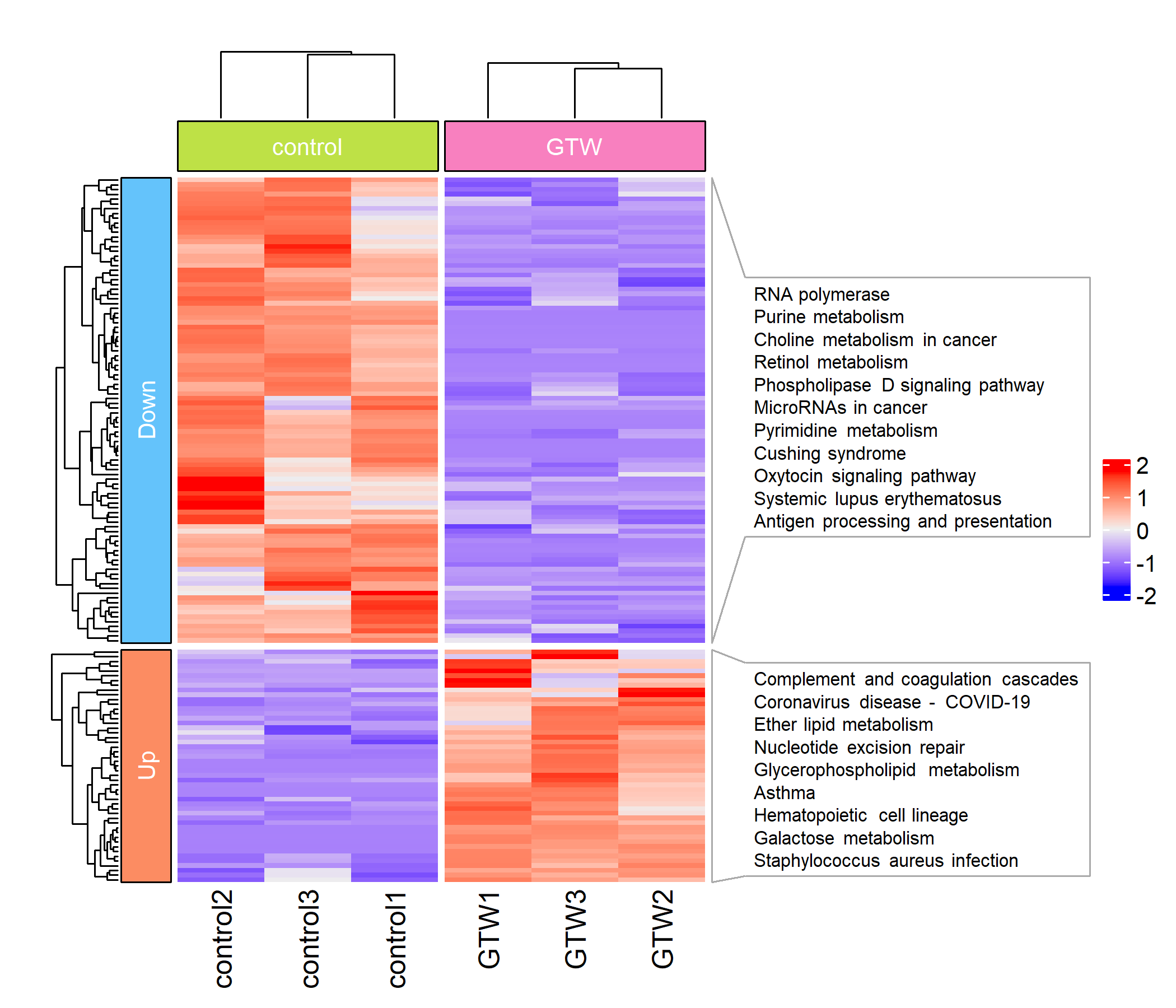
# 绘制热图
p = Heatmap(df.scale,
name = " ", # 热图的颜色图例名称
show_row_names = FALSE, # 不显示行名
show_column_names = TRUE, # 显示列名
row_split = dfGene$Group, # 按基因组分割行
column_split = dfSample$Group, # 按样本组分割列
cluster_row_slices = FALSE, # 不对行分割进行聚类
cluster_column_slices = FALSE, # 不对列分割进行聚类
column_title_gp = gpar(alpha = 0), # 设置列标题的透明度为0,即不显示列标题
row_title_gp = gpar(alpha = 0), # 设置行标题的透明度为0,即不显示行标题
top_annotation = HeatmapAnnotation(foo = anno_block(gp = gpar(fill = c("#bee146", "#f880bf")),
labels = unique(dfSample$Group), # 样本组标签
labels_gp = gpar(col = "white", fontsize = 10))), # 样本组标签样式
left_annotation = rowAnnotation(foo = anno_block(gp = gpar(fill = c("#64c3fb", "#fc8c62")),
labels = unique(dfGene$Group), # 基因组标签
labels_gp = gpar(col = "white", fontsize = 10))), # 基因组标签样式
right_annotation = rowAnnotation(
textbox = anno_textbox(
dfGene$Group, dfsentences, # 基因组与对应的富集条目
word_wrap = TRUE, # 自动换行
add_new_line = TRUE, # 添加换行符
background_gp = gpar(fill = "white", col = "#AAAAAA"), # 设置文本框背景样式
gp = gpar(col = "black", fontsize = 8)) # 设置文本颜色和字体大小
)
)
draw(p)
# 保存图片
# png("heatmap.png", width = 7, height = 6,res=300, units = "in") # 保存热图为PNG格式
# draw(p) # 绘制热图,并将图例放在右侧
# dev.off() # 关闭图形设备