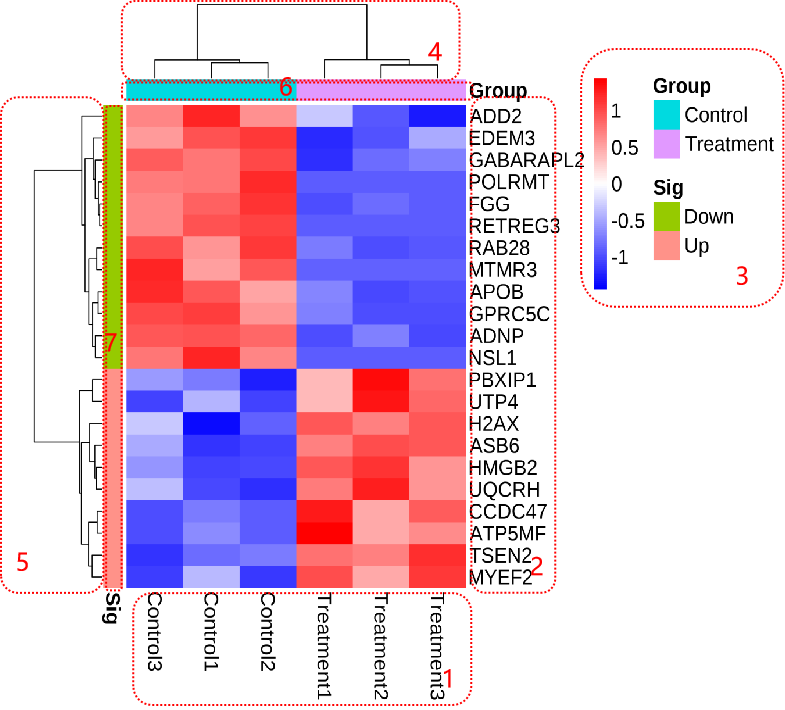
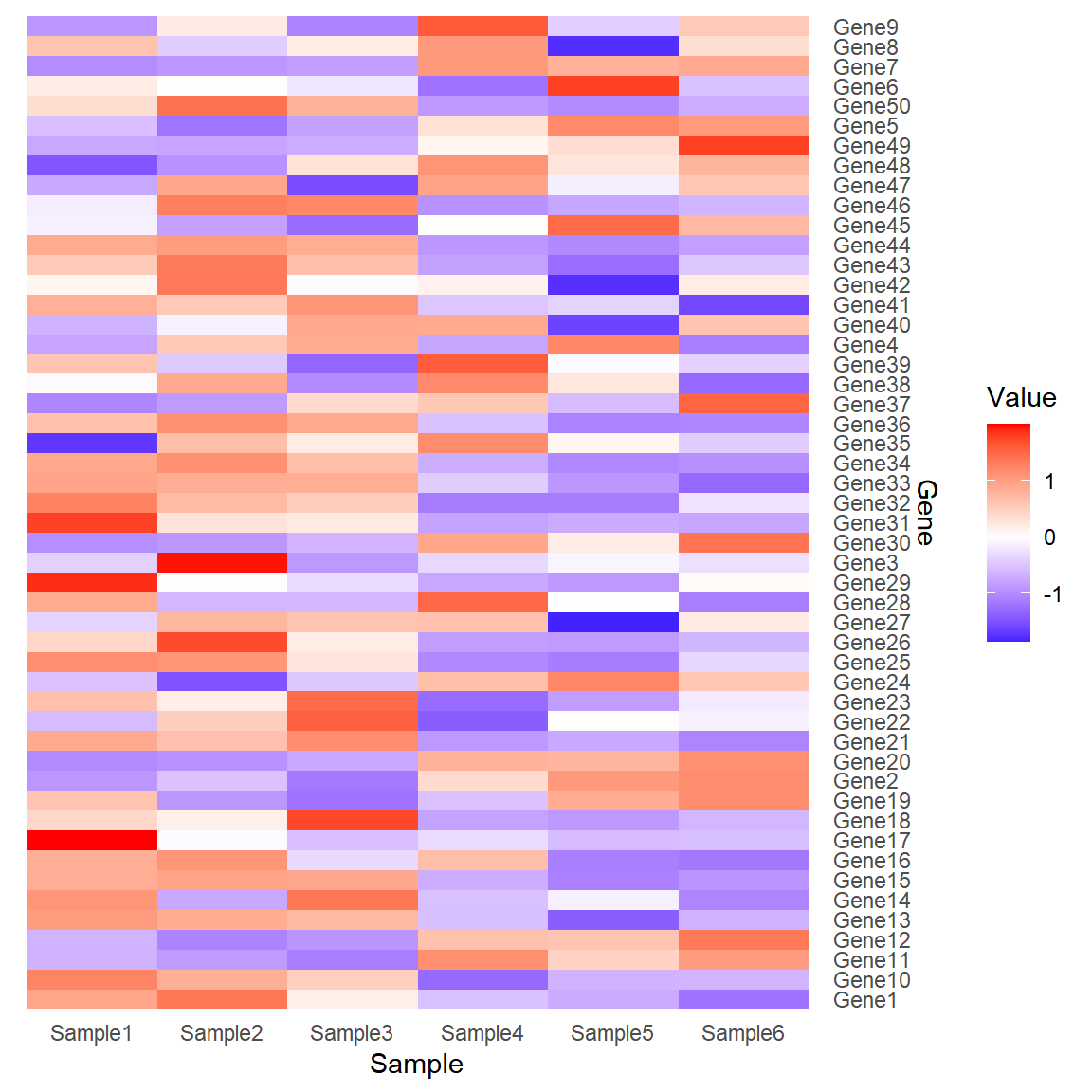
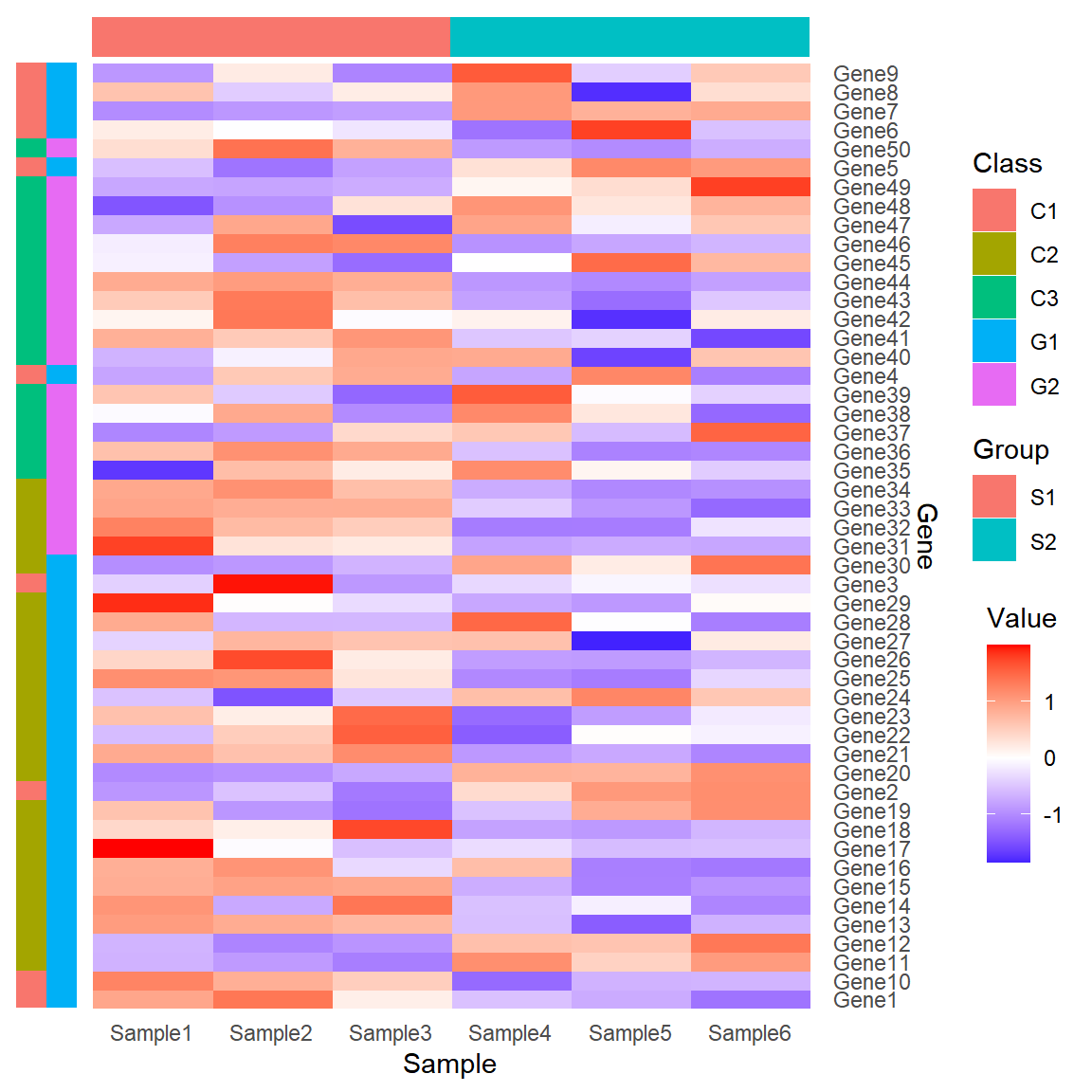
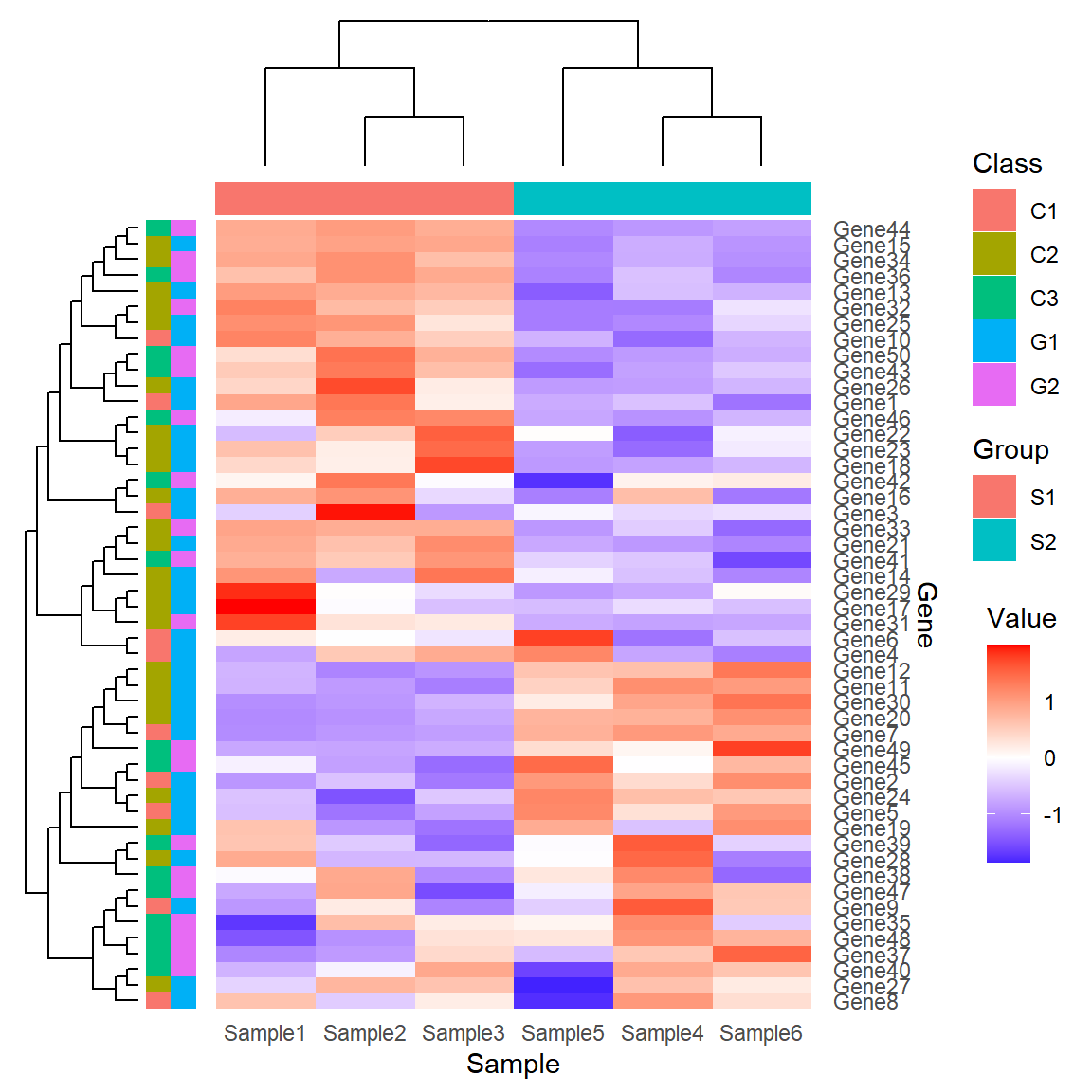
# 添加分组条带
# X轴(单条带例子)
dfSample = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/sample.class.txt")
pX = dfSample %>%
mutate(Y = "Group") %>%
ggplot(aes(x=X,y=Y,fill=Group))+
geom_tile() +
theme_void()+
labs(fill = "Group")
# y轴(双条带例子)
dfGene = read.delim("https://www.bioladder.cn/shiny/zyp/demoData/heatmap/gene.class.txt")
pY = dfGene %>%
pivot_longer(-1) %>%
ggplot(aes(x=name,y=X,fill=value))+
geom_tile() +
theme_void()+
labs(fill = "Class")
# 拼图
p %>%
insert_top(pX, height = .05)%>%
insert_left(pY, width = .09)