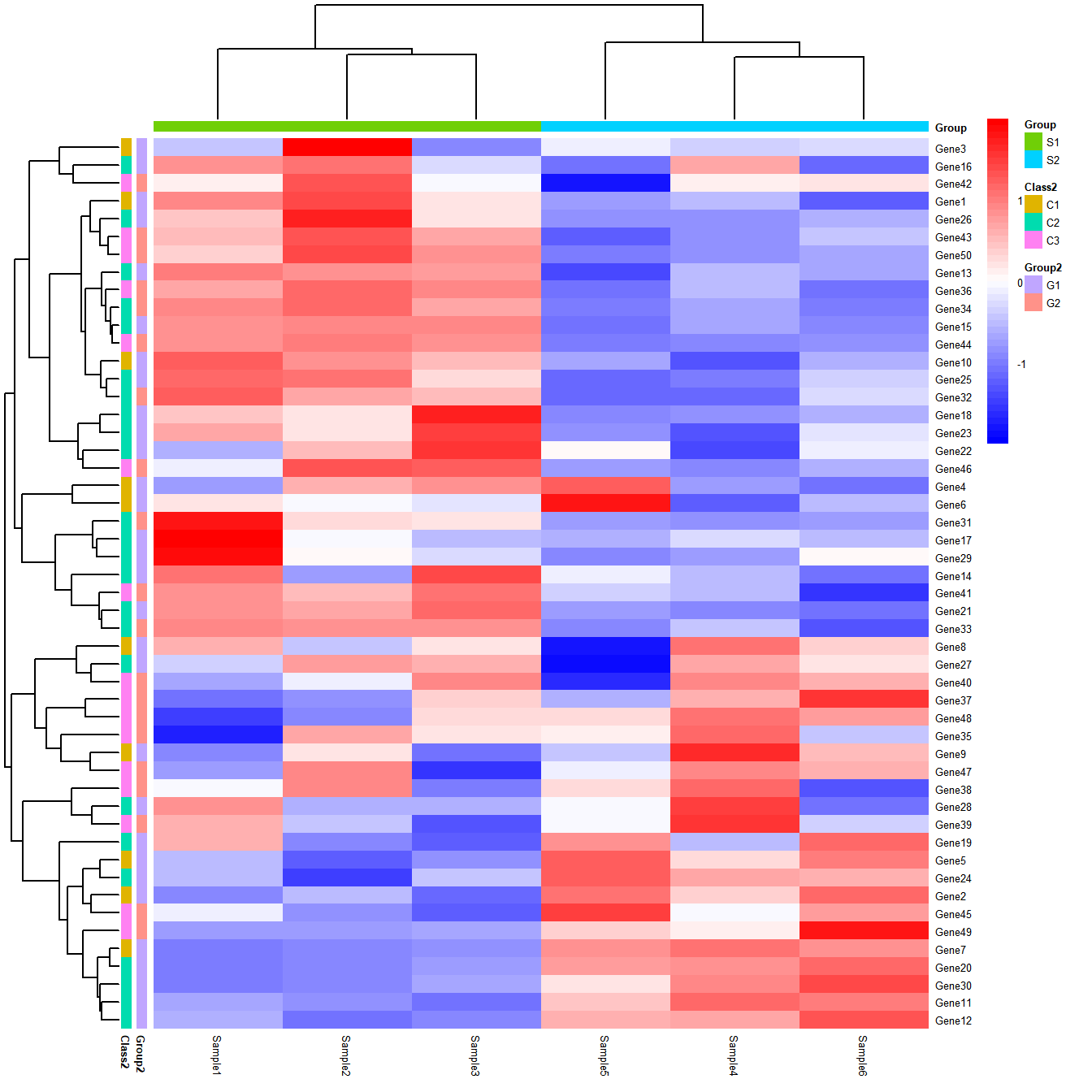
# 代码来源:https://www.r2omics.cn/
library(pheatmap) # 加载pheatmap这个R包
# 1,读取热图数据文件
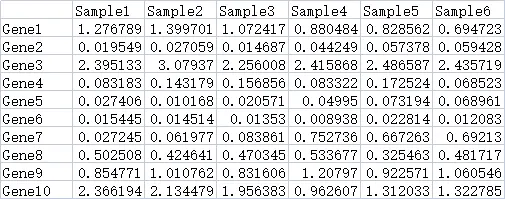
df = read.delim("https://www.r2omics.cn/res/demodata/heatmap/data.heatmap.txt", #文件名称 注意文件路径,格式
header = T, # 是否有标题
sep = "\t", # 分隔符是Tab键
row.names = 1, # 指定第一列是行名
fill=T) # 是否自动填充,一般选择是
# (可选)读取分组数据文件
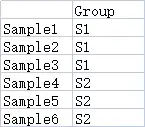
dfSample = read.delim("https://www.r2omics.cn/res/demodata/heatmap/sample.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
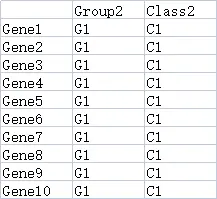
dfGene = read.delim("https://www.r2omics.cn/res/demodata/heatmap/gene.class.txt",header = T,row.names = 1,fill = T,sep = "\t")
# 2,绘图
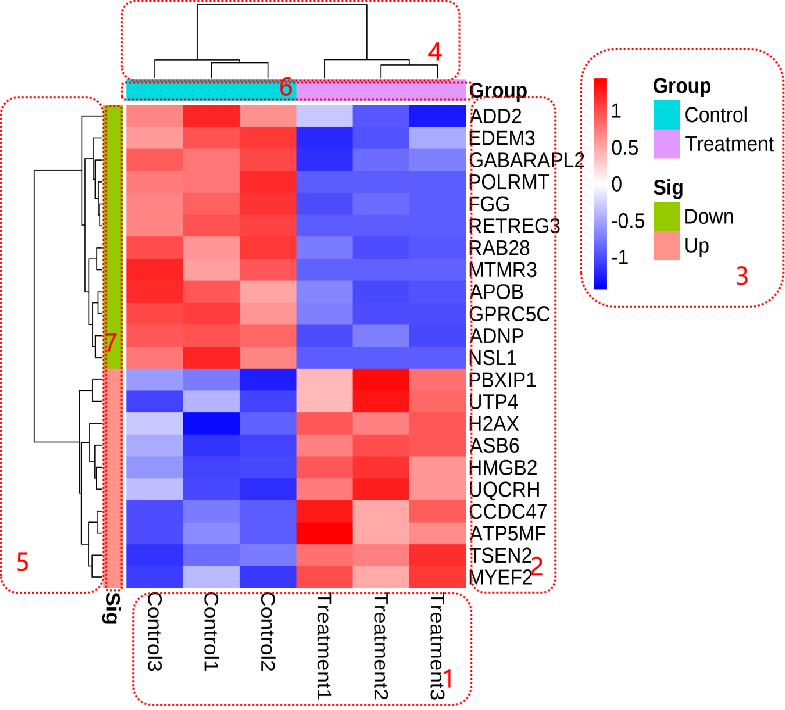
pheatmap(df,
annotation_row=dfGene, # (可选)指定行分组文件
annotation_col=dfSample, # (可选)指定列分组文件
show_colnames = TRUE, # 是否显示列名
show_rownames=TRUE, # 是否显示行名
fontsize=5, # 字体大小
color = colorRampPalette(c('#0000ff','#ffffff','#ff0000'))(50), # 指定热图的颜色
annotation_legend=TRUE, # 是否显示图例
border_color=NA, # 边框颜色 NA表示没有
scale="row", # 指定归一化的方式。"row"按行归一化,"column"按列归一化,"none"不处理
cluster_rows = TRUE, # 是否对行聚类
cluster_cols = TRUE # 是否对列聚类
)