# 代码来源:https://www.r2omics.cn/
# 加载R包,没有安装请先安装 install.packages("包名")
library(ggplot2)
library(ggbiplot)
# ggbiplot包需要从github上下载
# install.packages("devtools")
# library(devtools)
# install_github("vqv/ggbiplot")
# 读取PCA数据文件
df = read.delim("https://www.r2omics.cn/res/demodata/PCA/data.txt",# 这里读取了网络上的demo数据,将此处换成你自己电脑里的文件
header = T, # 指定第一行是列名
row.names = 1 # 指定第一列是行名
)
df=t(df) # 对数据进行转置,如果想对基因分组则不用转置
# 读取样本分组数据文件
dfGroup = read.delim("https://www.r2omics.cn/res/demodata/PCA/sample.class.txt",
header = T,
row.names = 1
)
# PCA计算
pca_result <- prcomp(df,
scale=T # 一个逻辑值,指示在进行分析之前是否应该将变量缩放到具有单位方差
)
# 绘图
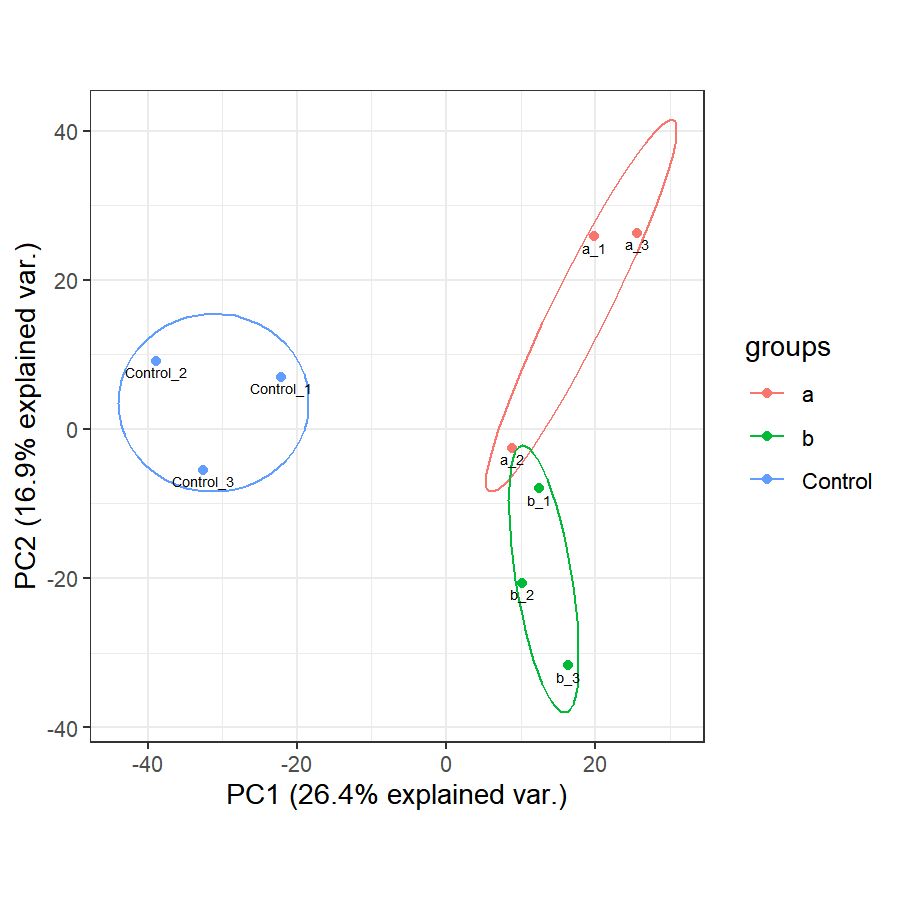
ggbiplot(pca_result,
var.axes=F, # 是否为变量画箭头
obs.scale = 1, # 横纵比例
groups = dfGroup[,1], # 添加分组信息,为分组文件的第一列
ellipse = T, # 是否围绕分组画椭圆
circle = F)+
geom_text( # geom_text一个在图中添加标注的函数
aes(label=rownames(df)), # 指定标注的内容为数据框df的行名
vjust=1.5, # 指定标记的位置,vjust=1.5 垂直向下1.5个距离。 负数为位置向上标记,正数为位置向下标记
size=2 # 标记大小
)+
theme_bw()